Capítulo 34 Agrupación
Los algoritmos que hemos descrito hasta ahora son ejemplos de un enfoque general denominado machine learning supervisado. El nombre proviene del hecho de que usamos los resultados en un set de entrenamiento para supervisar la creación de nuestro algoritmo de predicción. Hay otro subconjunto de machine learning denominado no supervisado. En este subconjunto, no necesariamente conocemos los resultados y, en cambio, estamos interesados en descubrir grupos. Estos algoritmos también se denominan algoritmos de agrupamiento (clustering en inglés) ya que los predictores se utilizan para definir grupos (clusters en inglés).
En los dos ejemplos que hemos utilizado en esta parte del libro, la agrupación no sería muy útil. En el primer ejemplo, si simplemente se nos dan las alturas, no podremos descubrir dos grupos, hombres y mujeres, porque la intersección es grande. En el segundo ejemplo, al graficar los predictores, podemos ver que descubrir los dos dígitos, 2 y 7, será retante:
library(tidyverse)
library(dslabs)
data("mnist_27")
mnist_27$train |> qplot(x_1, x_2, data = _)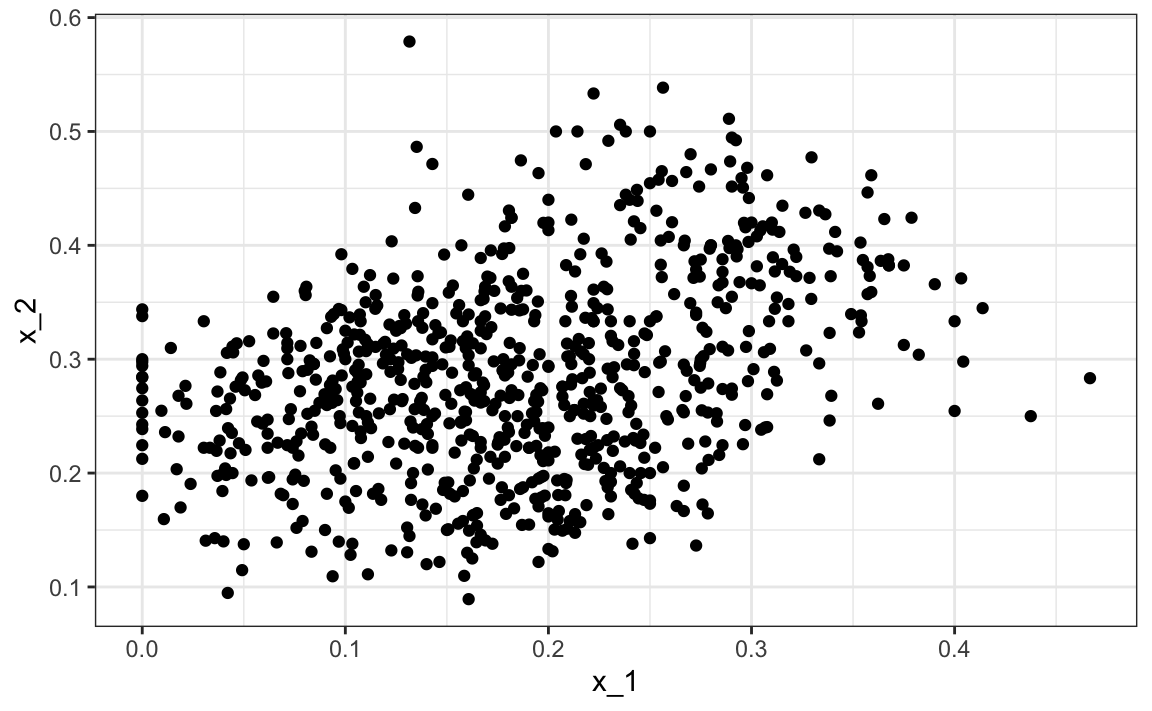
Sin embargo, hay aplicaciones en las que el aprendizaje no supervisado puede ser una técnica poderosa, en particular como una herramienta exploratoria.
Un primer paso en cualquier algoritmo de agrupamiento es definir una distancia entre observaciones o grupos de observaciones. Luego, decidimos cómo unir las observaciones en grupos. Hay muchos algoritmos para hacer esto. Aquí presentamos dos como ejemplos: jerárquico y k-means.
Construiremos un ejemplo sencillo basado en clasificaciones de películas. Aquí construimos rápidamente una matriz x que tiene calificaciones para las 50 películas con más calificaciones.
data("movielens")
top <- movielens |>
group_by(movieId) |>
summarize(n=n(), title = first(title)) |>
top_n(50, n) |>
pull(movieId)
x <- movielens |>
filter(movieId %in% top) |>
group_by(userId) |>
filter(n() >= 25) |>
ungroup() |>
select(title, userId, rating) |>
spread(userId, rating)
row_names <- str_remove(x$title, ": Episode") |> str_trunc(20)
x <- x[,-1] |> as.matrix()
x <- sweep(x, 2, colMeans(x, na.rm = TRUE))
x <- sweep(x, 1, rowMeans(x, na.rm = TRUE))
rownames(x) <- row_namesQueremos utilizar estos datos para averiguar si hay grupos de películas basados en las calificaciones de 139 calificadores de películas. Un primer paso es encontrar la distancia entre cada par de películas usando la función dist:
d <- dist(x)34.1 Agrupación jerárquica
Con la distancia calculada entre cada par de películas, necesitamos un algoritmo para definir grupos a partir de estas. La agrupación jerárquica comienza definiendo cada observación como un grupo separado. Entonces, los dos grupos más cercanos se unen en un grupo de forma iterativa hasta que solo haya un grupo que incluye todas las observaciones. La función hclust implementa este algoritmo y toma una distancia como entrada.
h <- hclust(d)Podemos ver los grupos resultantes usando un dendrograma.
plot(h, cex = 0.65, main = "", xlab = "")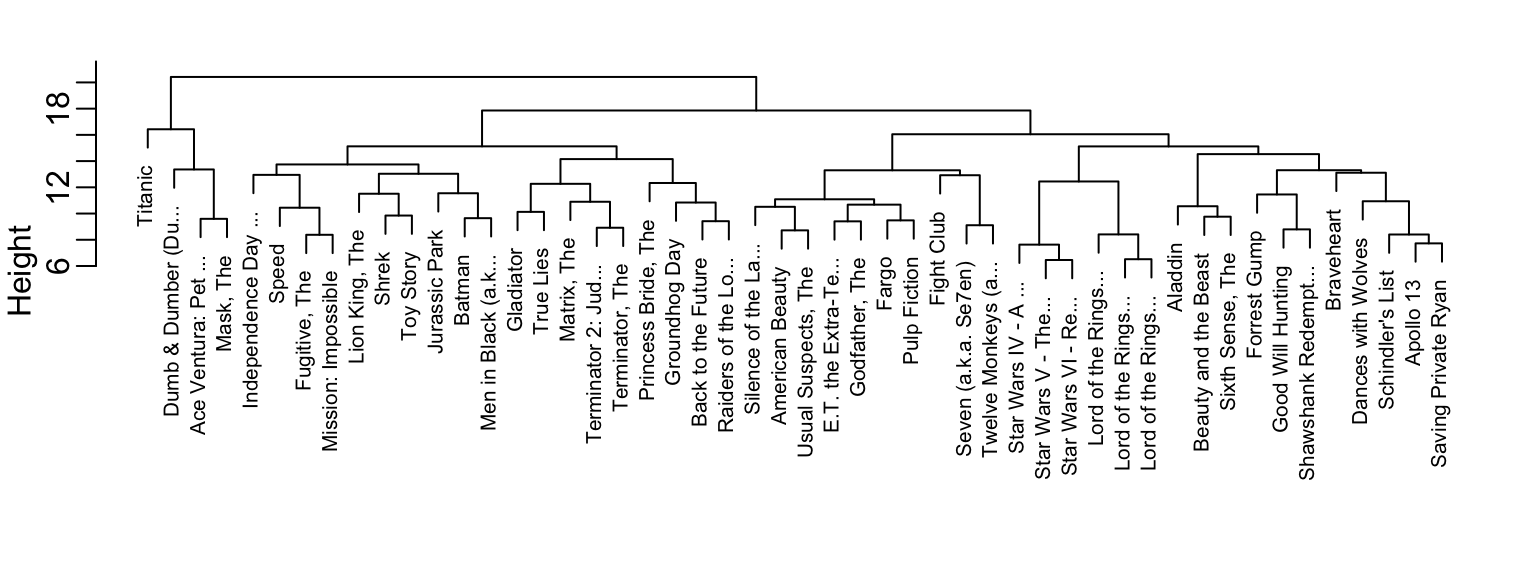
Para interpretar este gráfico, primero, determinamos la distancia entre dos películas encontrando la primera posición, de arriba a abajo, donde las películas se dividen en dos grupos diferentes. La altura de esta ubicación es la distancia entre estos dos grupos. Entonces, la distancia entre las películas de Star Wars es de 8 o menos, mientras que la distancia entre Raiders of the Lost of Ark y Silence of the Lambs es de aproximadamente 17.
Para generar grupos reales, podemos hacer una de dos cosas: 1) decidir la distancia mínima necesaria para que las observaciones estén en el mismo grupo o 2) decidir la cantidad de grupos que desean y luego encontrar la distancia mínima que lo logra. La función cutree se puede aplicar al resultado de hclust para realizar cualquiera de estas dos operaciones y generar grupos.
groups <- cutree(h, k = 10)Noten que la agrupación provee algunas ideas sobre los tipos de películas. El grupo 4 parece ser éxitos de taquilla:
names(groups)[groups==4]
#> [1] "Apollo 13" "Braveheart" "Dances with Wolves"
#> [4] "Forrest Gump" "Good Will Hunting" "Saving Private Ryan"
#> [7] "Schindler's List" "Shawshank Redempt..."Y el grupo 9 parece ser películas nerd:
names(groups)[groups==9]
#> [1] "Lord of the Rings..." "Lord of the Rings..." "Lord of the Rings..."
#> [4] "Star Wars IV - A ..." "Star Wars V - The..." "Star Wars VI - Re..."Podemos cambiar el tamaño del grupo haciendo k más grande o h más pequeño. También podemos explorar los datos para ver si hay grupos de evaluadores de películas.
h_2 <- dist(t(x)) |> hclust()34.2 k-means
Para usar el algoritmo de agrupamiento k-means, tenemos que predefinir \(k\), el número de grupos que queremos definir. El algoritmo k-means es iterativo. El primer paso es definir \(k\) centros. Luego, cada observación se asigna al grupo con el centro más cercano a esa observación. En un segundo paso, los centros se redefinen utilizando la observación en cada grupo: los medios de columna se utilizan para definir un centroide. Repetimos estos dos pasos hasta que los centros converjan.
La función kmeans incluida en base R no funciona con NAs. Con fines ilustrativos, reemplazaremos las NAs con 0s. En general, la decisión de cómo completar los datos que faltan, o si uno debería hacerlo, debe hacerse con cuidado.
x_0 <- x
x_0[is.na(x_0)] <- 0
k <- kmeans(x_0, centers = 10)Las asignaciones de grupos están en el componente cluster:
groups <- k$clusterRecuerden que debido a que el primer centro se elige al azar, los grupos finales son aleatorios. Imponemos cierta estabilidad al repetir la función entera varias veces y tomar el promedio de los resultados. El número de valores iniciales aleatorios para utilizar se puede asignar a través del argumento nstart.
k <- kmeans(x_0, centers = 10, nstart = 25)34.3 Mapas de calor
Una poderosa herramienta de visualización para descubrir grupos o patrones en sus datos es el mapa de calor (heatmap en inglés). La idea es sencilla: graficar una imagen de su matriz de datos con colores utilizados como señal visual y con tanto las columnas como las filas ordenadas según los resultados de un algoritmo de agrupamiento. Demostraremos esto con el set de datos tissue_gene_expression. Escalaremos las filas de la matriz de expresión génica.
El primer paso es calcular:
data("tissue_gene_expression")
x <- sweep(tissue_gene_expression$x, 2, colMeans(tissue_gene_expression$x))
h_1 <- hclust(dist(x))
h_2 <- hclust(dist(t(x)))Ahora podemos usar los resultados de esta agrupación para ordenar las filas y columnas.
image(x[h_1$order, h_2$order])Pero hay una función, heatmap, que lo hace por nosotros:
heatmap(x, col = RColorBrewer::brewer.pal(11, "Spectral"))No mostramos los resultados de la función heatmap porque hay demasiados atributos para que el gráfico sea útil. Por lo tanto, filtramos algunas columnas y rehacemos los gráficos.
34.4 Filtrando atributos
Si la información sobre los grupos se incluye en unos pocos atributos, incluir todos los atributos puede agregar suficiente ruido como para que detectar grupos sea retante. Un enfoque sencillo para tratar de eliminar atributos sin información es incluir solo aquellos con alta varianza. En el ejemplo de la película, un usuario con baja variación en sus calificaciones no es realmente informativo: todas las películas le parecen iguales. Aquí hay un ejemplo de cómo podemos incluir solo los atributos con alta varianza.
library(matrixStats)
sds <- colSds(x, na.rm = TRUE)
o <- order(sds, decreasing = TRUE)[1:25]
heatmap(x[,o], col = RColorBrewer::brewer.pal(11, "Spectral"))
34.5 Ejercicios
1. Cargue el set de datos tissue_gene_expression. Reste las medias de cada fila y calcule la distancia entre cada observación. Guarde el resultado en d.
2. Haga un gráfico de agrupamiento jerárquico y agregue los tipos de tejido como etiquetas.
3. Ejecute una agrupación k-means en los datos con \(K=7\). Haga una tabla que compara los grupos identificados con los tipos de tejidos correctos. Ejecute el algoritmo varias veces para ver cómo cambia la respuesta.
4. Seleccione los 50 genes más variables. Asegúrese de que las observaciones aparezcan en las columnas y que los predictores estén centrados. Agregue una barra de colores para mostrar los diferentes tipos de tejidos. Sugerencia: use el argumento ColSideColors para asignar colores. Además, use col = RColorBrewer::brewer.pal(11, "RdBu") para un mejor uso de los colores.